新規スピロ型キラル相間移動触媒を用いる
実用的アミノ酸合成
京都大学大学院 理学研究科 化学専攻 教授丸岡 啓二
1.はじめに
近年,地球規模で広がる環境への負荷をできるだけ軽減し,いわゆる環境に優しい化学 合成,環境に優しい分子・反応の設計を目指してより良い環境を作るためにグリーンケミス トリーへの取り組みが進んでいる。必要な物を望むだけ作ることを目指してきた有機合成化 学の分野でも,21世紀になると,資源を無駄遣いし環境汚染を広げてきた20世紀の化学 から質の転換が求められるのは致し方ないであろう。例えば,現在,縦横無尽の活躍をみせ ている有機金属触媒は有機物質変換の鍵を握る有用な機能性物質であり,その汎用性,多様 性の点から極めて優れた価値を有していることは言をまたない。ほんの10年ほど前には 見ることもなかったようなレアメタルを使う金属触媒が次々と開発され重要視されるに伴い, それらレアメタルの採取時と廃棄時の環境汚染がだんだんと問題になってくるであろう。 こういった観点から,私どもの研究室では金属を使わない環境調和型の不斉有機分子触媒 としてキラル相間移動触媒の開発に取り組んでいるが,その触媒設計と各種アミノ酸合成を はじめとする実用的不斉合成の最近の進展を紹介したい。2.相間移動触媒としての第四級アンモニウム塩
テトラアルキルアンモニウム塩 (R4N+X-)は,そのイオン構造のため通常水溶性であるが, そのアルキル基が長鎖になると脂溶性が高まり有機溶媒にも可溶となる。1) 1965 年頃,この ようなアルキルアンモニウム塩の特性を活かしたカルベンの付加反応に関する特許がStarks によって申請された。2) すなわち,濃い水酸化ナトリウム水溶液とクロロホルムから成る二 相溶媒系に,相間移動触媒としてトリオクチルメチルアンモニウムクロリドを加えると,こ の四級アンモニウム塩が水相と有機相の間を行き来する「相間移動触媒」として働き,オレ フィンのシクロプロパン化反応が速やかに進行するというものである。それまでは,この反 応の活性種であるジクロロカルベン1の発生には無水条件下,かなりの低温を必要とし,お のずから実験操作も繁雑であった。しかし,相間移動触媒を利用することにより,このよう な反応を常温でしかも水の存在下で行うことが可能となり,同時に反応速度の大幅な増大が 期待できるようになる。更に,実験操作が非常に簡便となるなど,様々な合成化学的利点か ら,その後の活発な研究につながっていった。3) CHCl3 Cl C Cl Cl Cl 1 60% (式 1) 触媒 NaOH 水溶液 触媒:トリオクチルメチルアンモニウム クロリド3.キラル相間移動触媒を用いる光学活性α - モノアルキルアミノ酸の合成
20世紀後半における有機合成化学の飛躍的な発展の中で,新たな反応性の獲得とともに, 反応の位置及び立体選択性の制御が極めて高いレベルで実現されるようになり,精密合成化 学という言葉が相応しい状況になっていった。その中でも触媒的不斉合成の進歩は目をみは るものがあり,真に実用的な不斉合成プロセスが開発されてきている。そのような背景のも と,近年,キラル相間移動触媒を用いた不斉合成反応の報告が幾つかなされてきたが実用的 見地からはほど遠く,最初の成功例は 1984 年にメルク社から報告された。4) すなわち, Dollingらはシンコニンアルカロイドとp-(トリフルオロメチル)ベンジルブロミドから得られ る光学活性第四級アンモニウムブロミド2をキラル相間移動触媒として用いることで,α -フェニルインダノン誘導体の不斉メチル化反応が高いエナンチオ選択性で進行することを見 い出した。この報告は,(i) 実用的不斉合成の最初の成功例であること;(ii) その後,開発さ れたキラル相間移動触媒は,ほとんどがシンコナアルカロイド由来である点で意義深い。 それから5年後の1989年,O’Donnellらはシンコニンから容易に合成可能なキラル相間移 動触媒3を用いることで,グリシンtert-ブチルエステルのベンゾフェノンシッフ塩基の不斉 アルキル化反応が温和な条件下で進行することを見い出した。5) ベンゾフェノンシッフ塩基 を用いることによりモノアルキル化が円滑に進行し, しかも生成したモノアルキル化体のα-プロトンはこの反応条件下で容易に脱プロトン化しないことが認められている。一方,シン コニジンから得られるキラル相間移動触媒4を用いると,逆の絶対配置をもったα - アルキ ルアミノ酸が得られてくる。このようにして得られるα - アルキルアミノ酸誘導体の光学収 率は必ずしも高くないが,酸処理でイミンとエステルの加水分解を同時に行った後,再結晶 でほぼ純粋な光学活性α-アミノ酸へと導くことができる。この報告を契機として,その後, 光学活性アミノ酸の不斉合成が幾つか報告されることになった。(式 3) まず,ImperialiやBowlerらは,シンコニジンをベンジル化したキラル相間移動触媒4を用 いると金属カチオンに対し,強い結合能力を有する非天然型アミノ酸,あるいはピリドキ サール補酵素を含むアミノ酸の不斉合成に有効であることを示した。6) (式 4,5) 50% NaOH N N H HO H H CH3-Cl 2 (10 mol%) O Cl Cl MeO CF3 O Cl Cl MeO CH3 N N H HO H H 20 °C, 18 h 95%, 92% ee 2 Br (式 2) キラル相間移動触媒 トルエン (シンコニンアルカロイド) キラル相間移動触媒,N Ph Ph O OBut N Ph Ph O OBut H H2N O OH H Cl Cl H 81% (66% ee) N N H HO H H 82% (62% ee) H [ >99% ee; ] 3 (10 mol%) CH2Cl2 20 °C, 12 h 50% NaOH N N H HO H H Cl 3 4 Cl CH2Br N Ph Ph O OBut H Cl Cl H2N O OH H Cl 4 (10 mol%) (式 3) 50% NaOH, CH2Cl2 [ >99% ee; ] 20 °C, 1 h N N Br N OBu t Ph Ph O N OBu t Ph Ph O N N H2N OH O N N 83% (53% ee) 6N HCl, 4 h 4 (20 mol%) (式 4) (式 5) 5 °C, 2 h 50% NaOH, CH2Cl2 N N OBu t Ph Ph O N OBut Ph Ph O 4 (20 mol%) [ >99% ee; ] CH3 Br O O N CH3 O O N H OH O N CH3 O O 68% (52% ee) Fmoc キラル相間移動触媒 キラル相間移動触媒 シンコニンアルカロイド由来の キラル相間移動触媒, シンコニジンアルカロイド由来の キラル相間移動触媒, 再結晶後 キラル相間移動触媒 キラル相間移動触媒 再結晶後 再結晶後
N H CO2H Fmoc CN N H CO2H Fmoc OMe OMe FmocHN N H O N N CO2H 7 5 6 さらに Imperiali らは,アミノ酸側鎖にそれ ぞれ蛍光を発する基,金属に配位可能な基や 蛍光消失基を導入することにより,5,6,7 のような非天然型アミノ酸を合成し,さらに 固相ペプチド合成法によりオリゴペプチドを 合成して,光励起による電子移動に基づく金 属イオンセンサーを作り上げている。7) HN O NH NH CONH2 HN O OH NH O Br O H2NOC N HN O NH O HO O HN CO2H OH O N NH O HN O OH NH NH HO Br O O OH Theonellamide F N H H N N H H N N H HN O H O OH OH OH O NH2 H O HO HO O O O H Sugar Cl O Sugar Cl HO O Sugar OH HO2C H Teicoplanin Rao や塩入らは,抗菌作用, 細胞毒性を示す環状ペプチド であるテオネラミドFの南半 球部の合成に,あるいはグリ コペプチド系抗生物質である テイコプラニンの不斉合成に, キラル相間移動触媒を用いる 不斉合成によって得られた光 学活性アミノ酸(点線で囲っ た部分)を利用している。9,10) O’Donnell らは,シンコナア ルカロイド由来のキラル相間 移動触媒8が塩基性条件下で どのように分解を起こすか詳 細に検討し,キラル相間移動 触媒が塩基性条件下で容易に O- アルキル化を起こし,得ら れた生成物9が活性な触媒に なることを見い出している。11) (式 7) (式 6) Pirrungらは,アミノ酸の側鎖にシクロオクタテトラエニル基を導入したアミノ酸を不斉合 成している。8) この場合,グリシンのアルキル化では相間移動反応を使っているものの不斉 導入はなされておらず,得られたモノアルキル化体を後に酵素による速度分割によって光学 活性α - アルキルアミノ酸誘導体を合成している。シクロオクタテトラエニル基を導入する ことによってペプチド鎖におけるアミドの回転と同様の構造変化が期待でき,また,電子移 動反応や遷移金属の配位子としても利用できる。 1) HCl 2) KOH 3) Ac2O N OMe Ph Ph O NH CO2H + Br Bu4N•HSO4 O N H CO2H O H2N CO2H 10% NaOH, CH2Cl2 20 °C, 24 h Acylase I
Corey らは,さらにこういったシンコニジンアルカロイドに構造的に剛直なアントラセン ユニットを組み入れることにより,新たなキラル相間移動触媒10 をデザインした。12) さら に,通常,用いられている有機溶媒/アルカリ水溶液の液/液系でなく,固体の水酸化セシ ウム・1水和物をジクロロメタン溶媒中で使う固/液系により,しかも低温下で反応を行な うことによりグリシンエステルの不斉アルキル化で高い不斉収率を得ている。 このキラル相間移動触媒10 は,グリシンエノラートのα , β - 不飽和カルボニル系への不 斉共役付加においても高いエナンチオ選択性が発現することが見い出されている。13) Coreyらと同じ時期に,Lygoらもシンコナアルカロイドにアントラセンユニットを導入す ることで,新たなキラル相間移動触媒11 や 12 を作り上げ,グリシンエステルの不斉アルキ ル化を液/液反応系で行うと高い不斉収率が得られることを見い出した。14) これらのアプ ローチは各種の天然及び非天然型α - アミノ酸合成に極めて有効であることを示している。 N Ph Ph O OBut N Ph Ph O OBut H H2N O OH H Ph Ph 10 (10 mol%) CsOH•H2O 84%, 94% ee PhCH2Br H N N H O H H + -78 °C, 23 h CH2Cl2 Br 10 N Ph Ph O OBut N Ph Ph O OBut H (CH2)2CO2Me 10 (10 mol%) CsOH•H2O 85%, 95% ee + -78 °C, 23 h CH2Cl2 OMe O N R1 N H HO H H 8 Br N R1 N H O H H OH N R1 N H R2O H H Br N R1 N R2O H H N R1 N H OH H R2-Br Base [R1 = CH 2Ph; CH2CH=CH2] 9 [R2 = CH 2Ph; CH2CH=CH2] (式 7) シンコニジンアルカロイド由来の キラル相間移動触媒, キラル相間移動触媒, キラル相間移動触媒 (式 8) (式 9) キラル相間移動触媒
特に,ジヒドロシンコニジンアルカロイドにアントラセンユニットを導入した触媒13(X = Cl)を用いると更に光学収率の向上(94% ee)が認められた。これらの報告を機に,相間移動 条件下での触媒的不斉合成の研究がいっそう加速されていくことになる。 N Ph Ph O OBut N Ph Ph O OBut H Ph N Ph Ph O OBut H Ph PhCH2Br 63% (89% ee) 68% (91% ee) 11 (10 mol%) 20 °C, 18 h 50% KOH / N N H HO H H N N H HO H H Cl 11 12 Cl 12 (10 mol%) N Ph Ph O OBut THF, N N H HO H H Br Br CO2But ButO 2C Ph2C=N N=CPh2 CO2But ButO 2C NH2 NH2 X OH O CO2H H2N NH2 NH2 HO HO CO2H CO2H NH2 CO2H 15% , 24 h 50% KOH, 13 55% ( ), 72% de, >95% ee 13 (10 mol%) こういったアントラセン メチル基を有するキラル相 間移動触媒13 (X = Br)は,ビ ス(α-アミノ酸)合成にも有 用であることが Ly g o らに よって報告された。15) ビス (α-アミノ酸)は,自然界で ジチロシン,イソジチロシ ンやメゾ - ジアミノピメリ ン酸などが知られているが, この方法を活用すると各種 の非天然型ビス(α-アミノ 酸)の合成が可能になる。 O’Donnell らは,不斉相間 移動反応プロセスにおいて 実用的見地からアルカリ水 溶液を用いる液/液系の反 応よりはイオン結合を持た シンコニンアルカロイド由来の キラル相間移動触媒, キラル相間移動触媒 シンコニジンアルカロイド由来の キラル相間移動触媒, キラル相間移動触媒 トルエン (式 10) (式 11) シンコニジンアルカロイド由来の キラル相間移動触媒, キラル相間移動触媒 くえん酸 室温 総収率 トルエン 室温 ジチロシン イソジチロシン
ない塩基としてホスファゼンを使って均一系の反応を行えば,大量合成にも適用できやすい ことを述べている。さらに,この系を使うと低温での反応が可能になるため,エナンチオ選 択性の向上が期待できる。16) また,O’Donnell らは,グリシンのエステル部を固相担持したものを用いて,非天然型α -アミノ酸の固相合成も行っている。17) このように,キラル相間移動触媒の適用範囲は多岐に わたっている。塩入らは,同様にシンコニンから誘導されるキラル相間移動触媒14 を用い て,さまざまな不斉合成反応の開発に成功している。18) その際,触媒の置換基 R として立 体的に嵩高いものを用いるのではなく,電子吸引性の置換基を導入するというアプローチで 触媒の修飾を行っている点は特に興味深い。これによって,四級中心である窒素原子上の電 子密度を下げ,反応基質から生成する対アニオンとの距離を縮めることで,反応の遷移状態 においてより強固な不斉場を形成できると考えられる。実際,ジブチルエーテル中,30%過 酸化水素水を酸化剤に,水酸化リチウムを固体塩基として用いた相間移動条件下でのα,β-不飽和ケトンの不斉エポキシ化反応では,触媒として14aを用いると生成物であるエポキシ ケトンにおいて不斉誘起はほとんど見られない(∼1% ee)。これに対して,キラル相間移動 触媒14b の存在下で反応を行った場合には,84% ee という高いエナンチオ選択性で生成物 が得られてくる。また,同様な相間移動条件下での不斉 Darzens 反応において,p-(トリフル オロメチル)基を有するキラル相間移動触媒14cを用いることで,α-ハロケトンとアルデヒ ドから光学活性エポキシケトンを満足のいく選択性で得ている。 N Ph Ph O OBut N Ph Ph O OBut H R R X
92% (94% ee) with MeI/BEMP 89% (89% ee) with EtI/BTPP 88% (91% ee) with PrI/BTPP 96% (90% ee) with Allyl-Br/BEMP 88% (91% ee) with BnBr/BEMP N P N NEt 2 N P N N N N Bu t But 10 (10 mol%) non-ionic base Me Me + -78~-50 °C CH2Cl2 BEMP BTPP N N H HO H H Ph Ph O Ph Ph O O Cl Ph O Ph O O CHO R Br 73%, 69% ee 14a : R = H 14b : R = I 14c : R = CF3 LiOH 14a: 72%, 1% ee 14b: 97%, 84% ee 30% H2O2 / Bu2O 4 °C 14 14c (10 mol%) + Bu2O, LiOH•H2O 4 °C 14 (5 mol%) キラル相間移動触媒 (式 12) (式 13) キラル相間移動触媒 キラル相間移動触媒 キラル相間移動触媒, ◎ 不斉エポキシ化反応 ◎ 不斉 Darzens 反応 キラル相間移動触媒, キラル相間移動触媒,
このように,キラル相間移動触媒の分野ではほとんどすべての系においてシンコナアルカ ロイド由来のキラル相間移動触媒が使われている。しかしながら,こういったアルカロイド から出発すれば触媒設計に限界があることは明白であり,通常は,(i) 第四級アンモニウム 塩合成のためのアルキルハライドを変える; (ii) シンコナアルカロイドの水酸基を保護する ためのアルキル基を変える,ぐらいしか方法がない。加えて,シンコナアルカロイド由来の キラル相間移動触媒は幾つかのβ - 水素をもっているため,アルカリ水溶液を加えた際, Hofmann脱離を引き起こし,触媒自体が分解してしまうといった欠点がある。こういった状 況下で我々は,(i) 合理的な触媒設計の観点から C 2対称軸を導入する;(ii) Hofmann 脱離を ひき起こすβ - 水素が不必要な系を構築する,という2大前提で次世代のキラル相間移動触 媒の創製に着手した。 まず,これらの条件を満たす単純な触媒として光学活性(S)-ビナフトール由来のキラルア ンモニウム塩15 を調製した。この触媒1モル%を用いてグリシン tert- ブチルエステルのベ ンゾフェノンイミンの不斉ベンジル化反応を検討した。トルエン/50%水酸化カリウム水溶 液中で相間移動反応を行なったところ,収率 34%で望ましいモノアルキル化体が得られ, その光学収率は 21%であった。光学活性ビス(α-ナフチル)アンモニウム塩15b を用いても 光学収率は 28%であった。こういった触媒15 ではエノラートが光学活性 ビナフチル部の 近傍にいる場合には不斉導入が期待できるが,アキラルなジベンジル位の部分では必然的 に光学収率の低下を招いてしまう。そこで更にジベンジル位の部分にもうひとつの光学活性 ビナフチル部を導入したスピロ型ビナフチルアンモニウム塩16aを調製すれば,より有効な 不斉環境が構築できる。この触媒は市販の光学活性 (S)- ビナフトールから出発して,無水ト リフルオロメタンスルホン酸/トリエチルアミンを用いてビス(トリフラート)に変換し,メ チルマグネシウムブロミドを用いたニッケル触媒によるクロスカップリング反応によって N Ph Ph O OBut N Ph Ph O OBut H Ph N Ar Ar + 15a, b (1 mol%) 50% KOH 0 °C 15a, b 15a (Ar = Ph) 15b (Ar = ) Br s PhCH2Br : 34% (21% ee) : 46% (28% ee) N Ph Ph N X X
[ X = halogen, OH, enolate ] [ ] 15a 16a キラル相間移動触媒 キラル相間移動触媒 キラル相間移動触媒 (式 14) アキラルな部分 α-Naph トルエン
ジメチル誘導体を得た。続いて,ベンゾイルパーオキシド/N- ブロモスクシンイミドを用 いるラジカルブロモ化によってジブロミドを得て,これをアリルアミンと反応させると環状 のアリルアミンが得られた。Wilkinson 触媒を用いる脱アリル化によって第2級環状アミン を得,これを先のジブロミドと反応させると望みのスピロ型ビナフチルアンモニウム塩16a へと導けた。 この触媒1モル%を用いてグリシン tert- ブチルエステルのベンゾフェノンイミンの不斉 ベンジル化反応を以前と同様の相間移動条件下で行うと,化学収率,光学収率ともに向上す ることが認められた。驚くべきことに,光学活性ビナフチル環の 3,3' 位にフェニル基を導入 した触媒16bを用いると,グリシンエステルの不斉アルキル化は更に速く進行することを見 い出し,これまで0℃で6時間要した反応がわずか30分後にはほぼ完結し,81%の収率で ベンジル化体が取れ,しかもその際の光学収率は 89%であった。さらに,3,3' 位をβ - ナフ チル基に変えたキラル触媒16cを1モル%用いることによって反応のエナンチオ選択性はさ らに向上し,わずか30分後には収率 95%,光学収率 96%になることが判った。19) OH OH Me Me OTf OTf Br Br NH N N Tf2O, Et3N CH2Cl2 MeMgBr NiCl2(PPh3)2 ether NBS (PhCO2)2 cyclohexane MeCN Allyl-NH2 RhCl(PPh3)3 MeCN-H2O MeOH K2CO3 Br 16a : 73%, 79% ee (R) for 6 h : 81%, 89% ee (R) for 0.5 h : 95%, 96% ee (R) for 0.5 h Br 16a (Ar = H) 16b (Ar = Ph) 16c (Ar = ) 16 N Ph Ph O OBut N Ph Ph O OBut H + 16a~c (1 mol%) 50% KOH 0 °C R PhCH2Br N Ar Ar Ph スピロ型のキラル相間移動触媒, キラル相間移動触媒 キラル相間移動触媒, キラル相間移動触媒 (式 16) (式 15) β-Naph トルエン
キラル相間移動触媒16 では,ふたつのビナフチル部は共に (S)- 異性体のものを使ってい るが,その一方を逆のエナンチオマーに変えるとどうなるだろうか? 試みにβ - ナフチル 置換されたビナフチル部に(R)-異性体のものを使ったキラル相間移動触媒17を作り上げ,そ れを1モル%用いて同様の相間移動条件下,グリシン tert- ブチルエステルのベンゾフェノ ンイミンの不斉ベンジル化反応を行なったところ,アルキル化反応は非常に遅くなり,しか もエナンチオ選択性も大幅に低下することが判った。 3,3' 位にβ - ナフチル基を導入したキラル相間移動触媒16c は,グリシン tert- ブチルエス テルのベンゾフェノンイミンの不斉アルキル化反応において高い一般性を有することがわか り,表1から明らかなように,わずか1モル%の触媒存在下,通常の相間移動反応条件下で 各種のアルキルハライドを用いるといずれの場合も90% ee以上の高いエナンチオ選択性が 認められた。19) 表1 . スピロ型キラル相間移動触媒を用いるグリシンエステルの不斉アルキル化反応 アルキルハライド 反応時間 エナンチオ選択性 収 率 PhCH2Br 0.5 h 96% ee 95% CH3I 8 h 90% ee 64% CH3CH2I 10 h 95% ee 41% CH2=CHCH2Br 1 h 94% ee 84% CH2=C(Me)CH2Br 1 h 93% ee 82% 1 h 95% ee 90% 0.5 h 96% ee 80% 1 h 96% ee 81% 1.5 h 96% ee 60% (注) 不斉アルキル化反応は,グリシンエステル(1当量)とアルキルハライド(1.2当量)をスピロ 型キラル相間移動触媒16cを1モル%存在下,トルエン/ 50%水酸化カリウム水溶液中,0℃で行 なった。 N Ph Ph O OBut N Ph Ph O OBut H Ph R 17 (1 mol%) 50% KOH 0 °C, 62 h N PhCH2Br + R S Br 17 53% (18% ee) Br Me Br F Br HC CCH2Br キラル相間移動触媒 トルエン キラル相間移動触媒, (式 17) β-Naph β-Naph
この触媒的不斉アルキル化反応の遷移状態を考察することにより,エナンチオ選択性発現 に関する知見を得ることができる。3,3' 位にβ - ナフチル基を導入したキラル相間移動触媒 16c の空間モデルを図1に示した。この図を見ると,置換されていない光学活性ビナフチル 部を土台に 3,3' 位にβ - ナフチル基を導入した光学活性ビナフチル部がその周囲を取り囲む ような形をとっており,そこにベンゾフェノンイミンのグリシン tert- ブチルエステルエノ ラートが近づくと無置換型ビナフチル部とベンゾフェノンイミン部との間で効率の良いπ, π -相互作用が働き,グリシン tert-ブチルエステルエノラートの一方のエナンチオ面が有効 に遮蔽されていることがわかる。この状態でアルキルハライドがエノラートに近づけば, 望ましい (R)- 配置を持ったα - アルキルアミノ酸が得られるという訳である。 図1 . キラル相間移動触媒16cを用いたグリシン誘導体の 触媒的不斉アルキル化反応の遷移状態図 さて,こういった相間移動反応では,反応スケールが大きくなればなるほど,反応の攪拌 効率が問題となってくる。特に二相の界面で反応が起こる場合,いかに効率良く撹拌するか が反応速度を大きく左右する。こういった問題も超音波照射を行なうことによって,ある程 度は解決できる。20) 例えば,単純なケトンのアルキル化反応を例に取ると,ベンジルフェニ ルケトンのベンジル化を通常の相間移動反応条件下,超音波照射すると,0℃,10分でア ルキル化体が定量的に得られるが,攪拌条件下ではわずか10%しか得られない。その他のメ チル化やブチル化でも,超音波照射と攪拌では反応性に大きな差が認められた。また,キラ ル相間移動触媒16c を用いるグリシン tert- ブチルエステルの不斉メチル化でも,通常の攪 拌条件では0℃,8時間で 64%の収率が得られるが,それを超音波照射すると,1時間後に は同様の収率が得られることが判る。その際,光学収率にほとんど差異は認められなかった。 N N Ph Ph O OBut R H N O O But R R-X
4 . キラル相間移動触媒を用いる光学活性α , α - ジアルキルアミノ酸の合成
光学活性α , α - ジアルキルアミノ酸は天然に存在しないものの,ペプチドの修飾や酵素 の阻害剤あるいは不斉合成における有用なキラル素子として高い潜在需要を持っている。従 来,Schollkopf,Seebach や Vedejs をはじめとする多くの化学者によって,光学活性α - モノ アルキルアミノ酸から化学量論的に各種のヘテロ環に変換し,それらのジアステレオ選択的 なアルキル化反応を行なうことによって,望みの光学活性α , α - ジアルキルアミノ酸18 へ と変換するのが通例であった。21) 1992 年になると,O’Donnell らは光学活性α , α - ジアルキルアミノ酸の触媒的合成法を報 告した。22) すなわち,シンコニンアルカロイド由来のベンジルアンモニウム塩3 を 10 モル %用いてアラニン tert- ブチルエステルの p- クロロベンズアルデヒドイミンの不斉アルキル 化反応を相間移動条件下で行うと,相当する光学活性α , α - ジアルキルアミノ酸19 が良い 収率で得られた。それらの光学収率はいずれの場合も50%以下と,それほど満足のいく値で はないが,光学活性α , α - ジアルキルアミノ酸合成の触媒反応例として評価できる。 Ph O Ph Ph O Ph R N Ph Ph O OBut N Ph Ph O OBut Me H BuI (5 eq); 15~25 °C, 2 h R-X = PhCH2Br (1.2 eq); 0 °C, 10 minMeI (5 eq); 15~25 °C, 30 min
(5 eq) 63%, 88% ee Bu4NBr (5 mol%) , R-X 50% KOH , 0 °C, 1 h Me-I + 16c (1 mol%) 50% KOH [0 °C, , 64%, 90% ee] : quant ( , 10%) : 85% ( , 2%) : 71% ( , 9%) E (Schollkopf) (Seebach) E N N Bz OM R Me O B N Me2N OM R Ph F H2N CO2H N N R OMe M MeO N N R OMe MeO (Vedejs) E * * E * H2N CO2H R E 18 N O OBut p-Cl-Ph N O OBut p-Cl-Ph Me Me R PhCH2Br R-X R-X = : 36% ee (78%) : 50% ee (84%) p-F-PhCH2Br CH2=CHCH2Br 3 (10 mol%) K2CO3/KOH, CH2Cl2 : 44% ee (80%) , 15 h 19 (式 18) 超音波照射: トルエン/ 水溶液 撹拌あるいは超音波 撹拌では 撹拌では 撹拌では 8 hの撹拌では 超音波 水溶液 キラル相間移動触媒 (式 19) 酸処理 キラル相間移動触媒 室温 (式 20) トルエン
- 続いて,Lygoらは自ら開発した,シンコニジンアルカロイドに構造的に剛直なアントラセ ンユニットを組み入れたキラル相間移動触媒12 を触媒量(10モル%)用いて光学活性α , α -ジアルキルアミノ酸19 の触媒的不斉合成を行なった。23) 不斉ベンジル化の場合は比較的 高い光学収率(77∼87% ee)が得られるが,その他のアルキルハライドでは選択性が著しく 低下してしまう。 こういった状況で,我々の研究室では最も効率の良い光学活性α , α - ジアルキルアミノ 酸の触媒的不斉合成プロセスの確立に取り組んだ。すなわち,グリシンから出発して,グリ シン tert- ブチルエステルのアルデヒドイミンに変換し,それをキラル相間移動触媒を用い た相間移動条件下,二種の異なるアルキルハライドを用いて同一容器内で連続的に不斉二重 アルキル化反応を行なおうというものである。得られたジアルキル化体は酸処理によって, 容易に望みの光学活性α , α - ジアルキルアミノ酸へと導ける。 そこで,3,3' 位にβ - ナフチル基を導入したキラル相間移動触媒16c を1モル%用い,ア ルキル化剤としてアリルブロミド,続いてベンジルブロミドを加えることによって,グリシ ン tert- ブチルエステルの p- クロロベンズアルデヒドイミンの不斉ジアルキル化反応をトル エン/ 50%水酸化カリウム水溶液中という相間移動条件下で行い,その後,生成物を 10% くえん酸で加水分解すると相当する光学活性α , α - ジアルキルアミノ酸エステル20 (R1 = アリル;R2 = ベンジル)が低収率ながらも良いエナンチオ選択性で得られた。アルキル化の 反応速度を上げるため,Corey らが使用した水酸化セシウムの水和物を用いる固/液反応系 を適用したところ,収率は 61%にまで向上し,またその際の光学収率も幾分良くなった。そ こで,さらにキラル相間移動触媒16 の 3,3' 位のアリール置換基の電子吸引,あるいは電子 供与効果を調べたところ,フルオロ置換基が反応性,選択性の点で顕著な効果を発現するこ とが認められ,特に3,4,5-トリフルオロフェニル置換基を有するキラル相間移動触媒21を用 いると,光学収率が 98%まで向上することを見いだした。24) H2N O OBut Me R N O OBut p-Cl-Ph N O OBut p-Cl-Ph 12 (10 mol%) Me Me 30 min R K2CO3/KOH, R-X AcOH THF-H2O : 87% ee (95%) R-X = PhCH2Br p-Cl-PhCH2Br n-Bu-I ICH2CO2But : 36% ee (low yield) : 19% ee (58%) : 77% ee (72%) 19 N O OBut p-Cl-Ph N O OBut p-Cl-Ph R1 R2 H2N O OH R1 R2 H+ H2NCH2CO2H 1) R1X, 2) R2X キラル相間移動触媒 室温, (式 21) (式 22) グリシン α,α- ジアルキルアミノ酸 キラル相間移動触媒 トルエン
その他の例を表2に示した。通常,反応性の高いアルキル化剤を使うと最初のアルキル化 は− 10℃,3時間半で終えることができ,続いて2番目のアルキル化剤を加えると0℃,1 時間以内でアルキル化がほぼ終り,加水分解後,相当する光学活性α , α - ジアルキルアミ ノ酸20 が良い収率で取れてくる。その際のエナンチオ選択性は∼ 98% ee にも達する。この 同一容器内での不斉ジアルキル化反応は中間体にキラルアンモニウムエノラート22 が生成 し,エノラート自体は完全な平面構造を有するため,最初のアルキル化で得られた不斉中心 は続くエノラート22 の生成で完全に消失してしまう。従って,このジアルキル化反応で得 られるエナンチオ選択性は,2番目の不斉アルキル化によってのみ決まることがわかる。 表2.スピロ型キラル相間移動触媒21を用いるグリシンエステル p- クロロベンズアルデ ヒドイミンの不斉ジアルキル化反応による光学活性α , α - ジアルキルアミノ酸 20の合成 アルキルハライド 最初の アルキルハライド 2番目の ( R1-X ) アルキル化条件 ( R2-X ) アルキル化条件 光学収率/収率 CH2=CHCH2Br -10 ℃, 3.5 h PhCH2Br 0 ℃, 0.5 h 98% ee 80% CH2=CHCH2Br -10 ℃, 3.5 h CH2=C(Me)CH2Br 0 ℃, 0.7 h 97% ee 60% CH2=CHCH2Br -10 ℃, 3.5 h HC≡CCH2Br 0 ℃, 0.5 h 96% ee 58% PhCH2Br -10 ℃, 3.5 h CH2=CHCH2Br 0 ℃, 0.3 h 92% ee 74% (注) 不斉ジアルキル化反応はスピロ型キラル相間移動触媒 21を1モル%の存在下,グリシン エステル(1当 量)にトルエン/水酸化セシウム水和物中,異なる2種のアルキルハライド(1∼1.2当量)を順次,加えることに よって行なった。 R1X = N O OBut p-Cl-Ph N O OBut p-Cl-Ph R1 R2 CsOH•H2O (5 eq) / -10~0 °C : 80%, 98% ee THF 21 CsOH•H2O (5 eq) / -10~0 °C : 28%, 83% ee 10% : 61%, 87% ee F F F F F F N Br 16c 50% KOH / 0 °C~r.t. H2N O OBut R1 R2 21 20 , R2X = PhCH 2Br (1 mol%) Br 1) R1X, 2) R2X キラル相間移動触媒 (式 23) トルエン溶媒/塩基 キラル相間移動触媒, キラル相間移動触媒, くえん酸 N O OBut p-Cl-Ph R1 NR*4 22
このため,光学活性α , α - ジアルキルアミノ酸合成において,出発物質をグリシンでなく α - アルキルアミノ酸にすると,中間体のエノラートはアキラルになるため,出発物質であ るα - アルキルアミノ酸は光学活性であるなしにかかわらず,不斉アルキル化によって,望 みの光学活性α , α - ジアルキルアミノ酸が得られることになる。例えば,アラニン,フェ ニルアラニンやバリンのエステルのp-クロロベンズアルデヒドイミンの不斉アルキル化をキ ラル相間移動触媒21(1モル%)の存在下で行なうと,高エナンチオ選択的に相当する光学 活性α , α - ジアルキルアミノ酸エステル23, 24 が得られる。24) N O OBut p-Cl-Ph H2N O OBut Me R Me N Br Boc 10% R-X PhCH2Br EtI CH2=CHCH2Br BrCH2CO2But 21 (1 mol%) 21 (1 mol%) N O OBut p-Cl-Ph H2N O OBut R1 R2 R1 10% THF : 73%, 98% ee : 78%, 91% ee R : 60%, 93% ee : 85%, 98% ee : 71%, 99% ee CsOH•H2O (5 eq) , -20~0 °C , -20~0 °C R1 = PhCH 2; R2 = CH2=CHCH2 R1 = i-Bu; R2 = PhCH 2 R1 = i-Bu; R2 = CH 2=CHCH2 : 71%, 97% ee : 64%, 92% ee : 70%, 93% ee R2-X THF CsOH•H2O (5 eq) (R1 = PhCH 2 or i-Bu) 23 24 RX = キラル相間移動触媒 くえん酸 キラル相間移動触媒 くえん酸 (式 24)
5.パーキンソン病の治療薬,
L- ドーパの化学合成
我々が本研究で編み出したスピロ型のキラル相間移動触媒16 や 21 は,C 2対称軸を有し ているため,出発となる光学活性ビナフトールを使い分けることによって,(S,S)型,(R,R)型 いずれのキラル相間移動触媒をも合成できるため,天然型,非天然型アミノ酸も含め,各種 のアミノ酸誘導体やそれらの関連体(アミノアルデヒド,アミノケトンやアミノアルコール など)の不斉合成に極めて有効であることがわかる。そういった合成的応用の一例として, L-ドーパエステルの不斉合成プロセスを次に示す。L-ドーパはパーキンソン病の治療に使わ れ,また,そのL-ドーパエステルはアルコール残基を変えることによって,いろんな薬理活 性を示すことが知られている。25) 従来,こういったL- ドーパエステルは,酵素法によって 合成されていた。すなわち,比較的高価なアミノ酸である光学活性チロシンから出発して, チロシナーゼを用いた酵素酸化によって L- ドーパのメチルエステルへと変換し,続いて各 種のアルコール存在下,α-キモトリプシンを用いてエステル交換を引き起こすことにより, 各種の L- ドーパエステルへと導かれている。こういった L- ドーパエステルもスピロ型の トルエン トルエンキラル相間移動触媒を使う化学合成によって,容易に得られるようになる。26) 例えば,(R)-ビナフトールから合成した (R,R)- 型のキラル相間移動触媒25 を1モル%存在下,アルキル 化剤として3,4-ビス(ベンジルオキシ)ベンジルブロミドを用い,グリシン tert-ブチルエステ ルのベンゾフェノンイミンの不斉アルキル化反応を相間移動条件下で行ない,得られたモノ アルキル化体のイミン部をくえん酸水溶液で加水分解することにより,β - ナフチル置換型 のキラル相間移動触媒25a の場合には,望ましいアルキル化体が 90% ee で得られるのに対 し,トリフルオロフェニル型のキラル相間移動触媒25b では 98% ee という高い光学収率が 得られた。アルキル化剤として4-(ベンジルオキシ)ベンジルブロミドを用いても同様の高い 光学収率が得られた。これらのモノアルキル化体は通常の接触水素添加反応を用いる脱ベン ジル化によって光学収率を損なうことなく,L-ドーパエステルやチロシンエステルへと変換 された。 H2N O OR H OH OH H2N O OH H OH OH H2N O OMe H OH H2N O OMe H OH OH ROH 0 °C, 1 h + (1.2 eq) 25 (1 mol%) 1 M THF : 80%, 90% ee with 25a : 81%, 98% ee with 25b N Ph Ph O OBut OBn Br R H2N O OBut H OBn R r.t., 10 h 50% KOH 25 H2N O OBut H OH R : 93%, 98% ee R = H : 83%, 98% ee with 25b 10% Pd/C, H2 THF, r.t., 5 h R = OH R = H : 94%, 98% ee [α]D29 = +19.7° (c 1.06, MeOH) [α]D29 = +26.1° (c 0.5, MeOH) R = OBn N Ar Ar 25a : Ar = 25b : Ar = 3,4,5-F3-Ph Br (式 25) (式 26) キラル相間移動触媒, キラル相間移動触媒 くえん酸 α- キモトリプシン [エステル交換] チロシナーゼ [酸化] L- ドーパ L- ドーパエステル 酵素法による合成 水溶液 トルエン β-Naph
6.おわりに
以上,私どもが現在取り組んでいるキラル相間移動触媒を用いる触媒的アミノ酸合成に関 する最近の進捗状況を紹介した。従来,汎用されてきたシンコナアルカロイド由来のキラル アンモニウム塩に較べ,我々の光学活性ビナフトール由来のスピロ型キラル相間移動触媒は, わずか1モル%で充分,アミノ酸の不斉アルキル化反応が行なえるという点は特筆すべきで あろう。このキラル触媒を用いて天然および非天然型の光学活性アミノ酸,あるいはその類 縁体の不斉合成研究をすすめていくにつれて,相間移動反応の化学に関していろいろな新し い知見が得られ,キラル触媒のさらなる改良に迫られている。特に,キラル相間移動触媒の 単純化に関する研究が興味深い局面を迎えており,そういった成果をこの紙上で紹介できな いのが残念である。 触媒的不斉合成の分野では,近年,不斉炭素中心構築のための方法論は著しい進歩を遂げ ている。その中でも繁雑な操作を必要としない相間移動条件下での触媒反応は,実用化が極 めて容易であり,工業的な面からも大いに注目されている。今後,相間移動条件下での高い 一般性と実用性を兼ね備えた不斉合成反応が次々と開発され,それらが医薬品に代表される 有用化合物の大量合成プロセスの確立に大きく寄与することが期待される。 最後に,本稿で述べられた研究成果は大井貴史助教授をはじめとする研究室の学生諸君の 献身的な努力の賜物であり,ここに心より感謝致します。 引用文献1) a) Makosza, M.; Ludwikow, M. Rocz. Chem. 1965, 39, 1223. b) Makosza, M.; Serafinowa, B.
Rocz. Chem. 1965, 39, 1401, 1595, 1647, 1799, 1805. c) Makosza, M. Pure Appl. Chem. 1975, 43, 439.
d) Dehmlow, E. V.; Dehmlow, S. S. Phase Transfer Catalysis, 3rd ed.; VCH: Weinheim, 1993. 2) a) Starks, C. M.; Napler, D. R. Fr. Demande, 1, 573, 164. b) Starks, C. M. J. Am. Chem. Soc. 1971, 93,
195.
3) a) Weber, W. P.; Gokel, G. W. 共著,田伏岩夫,西谷孝子共訳,「相間移動触媒」化学同人 (1978).
b) Phase-Transfer Catalysis ; Halpern, M. E. Ed., ACS; Washington (1997).
4) a) Dolling, U.-H.; Davis, P.; Grabowski, E. J. J. J. Am. Chem. Soc. 1984, 106, 446. b) Hughes, D. L.; Dolling, U.-H.; Ryan, K. M.; Schoenewaldt, E. F.; Grabowski, E. J. J. J. Org. Chem. 1987, 52, 4745. 5) O’Donnell, M. J.; Benett, W. D.; Wu. S. J. Am. Chem. Soc. 1989, 111, 2353.
6) a) Imperiali, B.; Prins, T. J.; Fisher, S. L. J. Org. Chem. 1993, 58, 1613. b) Imperiali, B.; Fisher, S. L.
J. Org. Chem. 1992, 57, 757. c) Kise Jr., K. J.; Bowler, B. E. Tetrahedron: Asymmetry 1998, 9, 3319.
d) Imperiali, B.; Roy, R. S. J. Am. Chem. Soc. 1994, 116, 12083. e) Imperiali, B.; Roy, R. S. J. Org. Chem.
1995, 60, 1891.
7) Torrado, A.; Imperiali, B. J. Org. Chem. 1996, 61, 8940. 8) Pirrung, M. C.; Krishnamurthy, N. J. Org. Chem. 1993, 58, 954.
9) Rao, A. V. R.; Reddy, K. L.; Rao, A. S.; Vittal, T. V. S. K.; Reddy, M. M.; Pathi, P. L. Tetrahedron Lett.
1996, 37, 3023.
10) Tohdo, K.; Hamada, Y.; Shioiri, T. Synlett 1994, 247.
11) O’Donnell, M. J.; Wu. S.; Huffman, J. C. Tetrahedron 1994, 50, 4507. 12) Corey, E. J.; Xu, F.; Noe, M. C. J. Am. Chem. Soc. 1997, 119, 12414.
13) Corey, E. J.; Noe, M. C.; Xu, F. Tetrahedron Lett. 1998, 39, 5347. 14) Lygo, B.; Wainwright, P. G. Tetrahedron Lett. 1997, 38, 8595.
15) a) Lygo, B.; Crosby, J.; Peterson, J. A. Tetrahedron Lett. 1999, 40, 1385. b) Lygo, B. Tetrahedron Lett.
1999, 40, 1389.
16) O’Donnell, M. J.; Delgado, F.; Hostettler, C.; Schwesinger, R. Tetrahedron Lett. 1998, 39, 8775. 17) O’Donnell, M. J.; Delgado, F.; Pottorf, R. S. Tetrahedron 1999, 55, 6347.
18) a) Arai, S.; Shioiri, T. Tetrahedron Lett. 1998, 39, 2145. b) Arai, S.; Tsuge, H.; Shioiri, T. Tetrahedron
Lett. 1998, 39, 7563.
19) Ooi, T.; Kameda, M.; Maruoka, K. J. Am. Chem. Soc. 1999, 121, 6519. 20) Ooi, T.; Tayama, E.; Doda, K.; Takeuchi, M.; Maruoka, K. Synlett 2000, 1500.
21) a) Duthaler, R. O. Tetrahedron 1994, 50, 1539. b) Seebach, D.; Sting, A. R.; Hoffmann, M. Angew. Chem.,
Int. Ed. Engl. 1996, 35, 2708. c) Wirth, T. Angew. Chem., Int. Ed. Engl. 1997, 36, 225. d) Cativiela, C.;
Diaz-de-Villegas, M. D. Tetrahedron: Asymmetry 1998, 9, 3517. 22) O’Donnell, M. J.; Wu, S. Tetrahedron Lett. 1992, 3, 591.
23) Lygo, B.; Crosby, J.; Peterson, J. A. Tetrahedron Lett. 1999, 40, 8671.
24) Ooi, T.; Takeuchi, M.; Kameda, M.; Maruoka, K. J. Am. Chem. Soc. 2000, 122, 5228.
25) a) Marrel, C.; Boss, G.; Van De Waterbeemd, H.; Testa, B.; Cooper, D. R.; Jenner, P.; Marsden, C. D.
Eur. J. Med. Chem. 1985, 20, 459. b) Cooper, D. R.; Marrel, C.; Van De Waterbeemd, H.; Testa, B.;
Jenner, P.; Marsden, C. D. J. Pharm. Pharmacol. 1987, 39, 635. c) Vulfson, E. N.; Ahmed, G.; Gill, I.; Goodenough, P. W.; Kozlov, I. A.; Law, B. A. Biotech. Lett. 1991, 13, 91. d) Ahmed, G.; Vulfson, E. N.
Biotech. Lett. 1994, 16, 367. e) Milewska, M. J.; Chimiak, A. Amino Acids 1994, 7, 89.
f) Brunner-Guenat, M.; Carrupt, P.-A.; Lisa, G.; Yeata, B.; Rose, S.; Thomas, K.; Jenner, P.; Ventura, P.
J. Pharm. Pharmacol. 1995, 47, 861.
26) Ooi, T.; Kameda, M.; Tannai, H.; Maruoka, K. Tetrahedron Lett. 2000, 41, 8339.
執筆者紹介 丸岡啓二(まるおか けいじ) 京都大学 大学院理学研究科 化学専攻 教授 [ご経歴] 1976年 京都大学工学部工業化学科卒業,1980年 ハワイ大学大学院化学科博士 課程修了,Ph. D. 取得。名古屋大学工学部応用化学科助手(1980∼1985),講師(1985∼ 1990),助教授(1990∼1995)を経て,1995年 北海道大学大学院理学研究科化学専攻教授, 2000年より現職。この間(2000∼2001)北海道大学大学院理学研究科化学専攻教授を併任。 昭和60年度,日本化学会進歩賞,及び平成12年度,井上学術賞受賞。 [ご専門] 有機合成化学,有機金属化学,分子認識化学,不斉合成化学,特に新しい概念 に基づく精密酸塩基触媒の創製と活用。
C0924 Chloromethylsulfonyl Chloride (McCl) inquire 飽和炭素上の求核置換反応は,最も基本的で広く研究されている有機反応の一つです。 この反応における脱離基としては,メシラート(Ms = CH3SO2)やトリフラート(Tf = CF3SO2)などが汎用されていますが,メシラートは反応性が弱くトリフラートは安定性が 低いため,天然物合成などの際に問題になることがあります。この問題を補える脱離基と して,クロロメタンスルホナート(モノクラート; Mc = ClCH2SO2)が清水,中田らによ り最近見出されました。 本品1は,塩化メチレン溶媒中ピリジンなどの塩基存在下0℃でアルコールと反応させ ることにより,相当するモノクラートをほぼ定量的に与えます。多くの場合,トリフルオ ロメタンスルホニル化と同様に容易にクロロメタンスルホニル化は進行し,メシル化より も早い速度で完了します。また,得られたモノクラートは精製することなく,そのまま次 の反応に利用することができます。 脱離基としてモノクラートを用いることにより,第二アルコールの立体反転,炭素−炭 素結合の転位,環状エーテルの環拡大などの有用な反応を行うことができます。 第二アルコールの立体反転は,アセテートを経由して行われます1)。ステロイドのモノ クラート 2a にベンゼン溶媒中18-クラウン-6 の存在下,酢酸セシウムを作用させ加熱還流 することにより,立体反転したアセテートが良好な収率で得られます。これを加水分解あ るいは還元することによりほぼ定量的に相当する第二アルコールが生成します。メシラー ト 2b を用いた場合には反応速度が非常に遅く,収率も劣っています。トリフラート 2c は 不安定で単離できません。
Yield from Alcohol
Reaction Time Acetate Olefins
2a 5 h 93% 4%
2b 4 d 82% 6%
2c (2c was not isolated)
一方,C-3位の水酸基に隣接したC-4位に2つのメチル基を置換した3β-hydroxygrindelate をモノクラート 3a にした後,酢酸亜鉛あるいは酢酸セシウムを作用させると炭素−炭素 結合の転位反応が起こります2)。モノクラート3aに上記と同様の反応条件で酢酸セシウム を反応させると立体反転したアセテート 4 は得られずに,4α-メチル基が3位に転位した R1 R2 OH R1 R2 OMc McCl , Pyridine 1 0 °C ~ r.t. Me Me Me Me H RO Me Me H AcO base or LAH CsOAc, 18-crown-6 benzene, reflux 2a: R=Mc 2b: R=Ms 2c: R=Tf Me H HO
5が主生成物として得られます。これは,立体的に込み合っている C-3 位をアセテートア ニオンが攻撃できないためであると考えられています。転位生成物 5 は,ジオキサン中 90 ℃で酢酸亜鉛を反応させる条件において最も収率良く得られます。
Reaction Condition 5 (Yield from Alcohol)
3a CsOAc, 18-crown-6, benzene, reflux, 3 d 69%
3a Zn(OAc)2, dioxane, 90 ℃, 3 d 75%
3b Zn(OAc)2, dioxane, 90 ℃, 3 d 8%
3c (3c was not isolated and 5 was directly obtained) 6%
また,モノクラートは環状エーテルの環拡大反応にも利用されます。テトラヒドロフラ ン化合物から誘導されたモノクラート 6a を酢酸−水(1:1)混合溶媒中,酢酸亜鉛存在下 50 ℃で加熱することにより,2,3-trans- テトラヒドロピラン7a, b が良好な収率で得られま す3)。メシラート 6b を用いて同様の条件下で反応を行うと未反応の 6b が回収されてしま い,効率良く環拡大反応を行うには加熱還流する必要があります。また,モノクラート 6a はジオキサン−水(1:1)混合溶媒を用いる中性条件下においても収率良く7aを与えるため, 酸や塩基に弱い官能基を有する環状エーテルの合成に有効です。このモノクラートを利用 した環拡大反応は,Hemibrevetoxin B 全合成における CD 環の合成に利用されています4)。
Reaction Condition 7 (Yield from Alcohol)
6a Zn(OAc)2, AcOH-H2O, 50 ℃, 4 h 85% (7a: 77%, 7b: 8%)
6a Zn(OAc)2, dioxane-H2O, 50 ℃, 6 h 97% (7a)
6b Zn(OAc)2, AcOH-H2O, 50 ℃, 24 h 13% (7a) (6b: 67%)
モノクラートを脱離基として用いる方法は,これらの他にも第二アルコールの立体反転
を伴うアジドへの変換反応や第一アルコールのニトリルへの変換反応などにも利用されて います2)。
文 献 1)Efficient method for inversion of secondary alcohols
T. Shimizu, S. Hiranuma, T. Nakata, Tetrahedron Lett., 37, 6145 (1996); 38, 3655 (1997). 2)Rearrangement of the C-C bond and conversion of alcohols into azides and nitriles
T. Shimizu, T. Ohzeki, K. Hiramoto, N. Hori, T. Nakata, Synthesis, 1999, 1373. 3)Rearrangement-ring expansion of cyclic ethers
N. Hori, K. Nagasawa, T. Shimizu, T. Nakata, Tetrahedron Lett., 40, 2145 (1999). 4)Total synthesis of hemibrevetoxine B
M. Morimoto, H. Matsukura, T. Nakata, Tetrahedron Lett., 37, 6365 (1996). Me O CO2Me Me Me H Me Me RO Me H Me CH2 Me H AcO Me Me AcO AcO 3a: R=Mc 3b: R=Ms 3c: R=Tf 4 5 3 4 5 O OAc O OR 1 OR2 Me H Me OR AcO 7a: R1=Ac, R2=H 7b: R1=R2=Ac 7c: R1=R2=H 6a: R=Mc 6b: R=Ms
D2755 5,5'-Dibromo-2,2'-bithiophene 5g 21,400 円 1g 7,250 円 S S Br Br + S MgBr Ni(dppp)Cl2 S S S S Ni(dppp)Cl2 S S S S S S C8H17 88% C8H17 C8H17 NBS 81% S S S S C8H17 C8H17 Br Br 84% S MgBr C8H17 C8H17 C8H17 C8H17 C8H17 1 S S S S S S C8H17 C8H17 C8H17 C8H17 S S S S S S H C8H17 C8H17 S C8H17 C8H17 n n=1~6 長鎖共役分子は導電材料や EL 材料,非線型材料など,多彩な先端機能材料として注目 されています。この長鎖共役分子の中でもオリゴチオフェン類は,架橋部にS原子がある ことによる構造的安定性や,側鎖や置換基を導入しやすい点などから,活発に研究されて います1)。 大坪らは本品 1 を用いて長鎖オリゴチオフェンの合成を行い ,単一分子共役系として は最長となる 48 量体の合成に成功しました1b,2)。 最近では末端にフラーレンやポルフィリンを修飾したオリゴチオフェンによる,電子や エネルギー伝達系の研究も盛んに行われています3)。 文 献 1)Review
a) R. E. Martin, F. Diederich, Angew. Chem. Int. Ed., 38, 1350 (1999). b)大坪徹夫 , 安蘇芳雄 , ファインケミカル , 29(10), 5 (2000).
2)Synthesis and properties of the longest oligothiophenes
H. Nakanishi, N. Sumi, Y. Aso, T. Otsubo, J. Org. Chem., 63, 8632 (1998). 3)Intramolecular energy transfer of [60]fullerene-linked oligothiophenes
T. Yamashiro, Y. Aso, T. Otsubo, H. Tang, Y. Harima, K. Yamashita, Chem. Lett.,
1999, 443. 関連製品 B1276 2,2'-Bithiophene 10g 16,600円 1g 3,300円 B1838 2,2'-Bithiophene-5-carboxaldehyde 5g 17,900円 1g 6,850円 B1874 5-Bromo-2,2'-bithiophene-5'-carboxaldehyde 5g 24,600円 1g 8,150円 B2058 5''-Bromo-2,2':5',2''-terthiophene-5-carboxaldehyde 1g 34,100円 100mg 6,150円 T1196 2,2':5',2''-Terthiophene (=α-Terthienyl) 5g 46,500円 1g 12,200円 T1805 2,2':5',2''-Terthiophene-5-carboxaldehyde 1g 19,500円 B1067 3-Bromothiophene 250g 48,200円 25g 8,250円
B2189 4'-Bromobenzo-15-crown 5-ether 1a 5g 46,900 円 1g 13,700 円 B2181 4'-Bromobenzo-18-crown 6-ether 1b 5g 29,800 円 1g 8,600 円 O O O O O Br 1a: n=1 1b: n=2 O O O O O PPh2 O O O O O CHO O O O O O CH O O O O O S CO2-Cs+ CO2-Cs+ i) nBuLi ii) Ph2PCl Pd(dba)2 N-ethylpiperidine 2 O O O O O S i) nBuLi ii) DMF iii) HCl S Bu3Sn 10 mol% Pd(PPh3)4 dba=dibenzylideneacetone 1) 2) 3) 4) CHCOOH CH2 CHCOOH n n n 2 3 2 n カチオン捕捉能を有するクラウンエーテル類と種々の機能を有する化合物を結合させる ことで,新しい機能や優れた能力を引き出す試みがなされています。例えば,本品 1 より 誘導される2は,クラウンエーテルとトリフェニルホスフィンを組み合わせた構造を有し ています。2 を配位子としたパラジウム錯体は,二相系での NaCN を用いたアリールハラ イドのシアノ化反応において優れた触媒として働きます1)。また,アリールチオフェン蛍 光団の構造を組み合わせた 3 は,Na+および K+に対する蛍光指示薬として合成されてい ます2)。従って,本品 1 は機能を付加させたベンゾクラウンエーテル類の合成に,有用な 原料として用いられます。
文 献 1)Catalytic cyanation of aryl halides with NaCN
T. Okano, M. Iwahara, H. Konishi, J. Kiji, J. Organomet. Chem., 346, 267 (1988). T. Okano, M. Iwahara, J. Kiji, Synlett, 1998, 243.
2)Fluorescent indicators for Na+ and K+
E. Cielen, A. Tahri, K. V. Heyen, G. J. Hoornaert, F. C. De Schryver, N. Boens,
J. Chem. Soc., Perkin Trans. 2, 1998, 1573.
3)Preparations of alkenylbenzocrown ethers
K. Kikukawa, S. Takamura, H. Hirayama, H. Namiki, F. Wada, T. Matsuda, Chem. Lett.,
1980, 511.
4)Preparations of formylbenzocrown ethers
S. P. Gromov, O. A. Fedorova, A. I. Vedernikov, V. V. Samoshin, M. V. Alfimov,
Russ. Chem. Bull., 42, 960 (1993).
S. P. Gromov, O. A. Fedorova, A. I. Vedernikov, V. V. Samoshin, N. S. Zefirov, M. V. Alfimov, ibid., 44, 116 (1995).
B2128 2-(4-Bromobutyl)-1,3-dioxolane 1 5g 20,200 円 C1678 2-(2-Chloroethoxy)tetrahydro-2H-pyran 2 25g 8,400 円 本品1および2のような片側を保護した二官能性化合物は,様々な有機合成反応に使用 されています。例えば,ホルミル基を保護した 1 は,分子内 Wittig 反応を経由した大環状 ラクトンの合成に使用されています1)。また,水酸基を保護した 2 は,アザクラウンエー テル類の合成に利用されています2)。1 および 2 の 1,3- ジオキソランとテトラヒドロピラ ニルエーテルは,アルカリ性条件下で安定に存在し,酸性条件下において脱保護されます。 文 献 1)Synthesis of (±)-Recifeiolid
H. J. Bestmann, R. Schobert, Synthesis, 1989, 419.
2)Synthesis of 8-hydroxymethyl-4,7,10,13-tetraoxa-1-azacyclopentadecane
B. Son, B. Czech, R. A. Bartsch, Synthesis, 1984, 776.
関連製品 A1176 2-(2-Aminoethyl)-1,3-dioxolane 5g 18,800円 1g 7,250円 A1179 2-(Aminomethyl)-1,3-dioxolane 1g 12,500円 B1132 2-(2-Bromoethyl)-1,3-dioxolane 250g 43,500円 25g 8,400円 B1152 2-Bromomethyl-1,3-dioxolane 25g 8,600円 C1321 2-Chloromethyl-1,3-dioxolane 25g 4,000円 B1182 2-(2-Bromoethyl)-1,3-dioxane 250g 55,000円 25g 9,300円 C1279 2-(4-Chlorobutoxy)tetrahydropyran 5g 4,900円 V0076 2-Vinyloxytetrahydropyran 5g 8,400円 O O Cl O O BnO OH OH NaH O O BnO O O OH OH O O HO O O NH Br O O OH O PPh3 O O O O O O C C PPh3 i) ii) HCl 4 2 , i) LiNH2 , ii) Na / liq. NH3 iii) 0.1N HCl 1 3 1) 2) CO2H Cl CO2H Cl CO2H Cl CO2H Cl CO2Me Cl N H CO2Et Cl O
Chiral Building Blocks
C1373 5g 19,100円 (R)-(+)-C1633 5g 8,600円 C1377 5g 32,700円 (R)-(+)-B2141 100mg 2,750円 1g 6,350円 (S)-(–)-C1634 5g 9,500円 1g 11,100 円 (S)-(–)-B2142 100mg 4,200円 HO OBn OH C1372 5g 19,100円 C1371 5g 21,200円 E0533 5g 14,500円 (R)-(–)-N0655 5g 24,300円 1g 7,350円 1g 7,050円 (S)-(+)-N0679 5g 24,300円
分析項目 ●有機元素分析 (C,H,N,O,S) (N) ●赤外吸収スペクトル (FT-IR) ●核磁気共鳴分析 (NMR) ●ガスクロマトグラフ分析 (GC) ●ガスクロマトグラフ・質量分析 (GC-MS) ●紫外・可視分光分析 (UV, VIS) ●液体クロマトグラフ分析 (HPLC) ● ICP 発光分光分析 ●滴定 ●その他 D2831 2,2-Difluoro-1,3-dimethylimidazolidine 25g 27,300 円 5g 8,400 円 医薬品などの生理活性物質の特定位置にフッ素原子を導入すると,薬効が向上したり, 毒性が軽減されることがあります。そのため,生理活性物質の特定位置にフッ素を導入 する方法が盛んに研究されています。含フッ素有機化合物は天然にはほとんど存在せず, 合成のある段階でフッ素化が必要です。 本品 1 は熱安定性に優れたフッ素化剤で,アルコール,カルボン酸,アルデヒド,ケト ン等と反応し,フッ素化合物を生成します。また,n-へキサンから非プロトン性極性溶媒 までの幅広い溶媒と相互に溶解します。そのため,幅広い応用性を持つフッ素化剤として 期待されています。 文 献 1) Chem. Abstr., 132, 137116. 2) Chem. Abstr., 133, 58608. DMI, 2 °C, 10 min 1 N N Me Me F F CH2OH CHOH CH2OH CH3 C O CH2F Y.90% 1) DMI = 1,3-Dimethyl-2-imidazolidinone MeCN, reflux, 5 h S O OK KO S O F F 1 O O 2) Y.70% お知らせ ●微細試料のIRスペクトル が取れ るようになりました。異物の同定な どにご利用ください。 ●マクロ窒素分析 ができるように なりました。ケルダール法の代わり にご利用ください。 東京化成工業株式会社 分析センター 〒 114-0003 東京都北区豊島 6-15-9 TEL 03-3919-5131 FAX 03-3919-7156 E-mail : [email protected] http://www.tokyokasei.co.jp/bunseki/
17,000 品目を超える有機試薬で培った確かな分析技術
キラル磁気異方性試薬
/ Chiral Derivatizing Agent for Absolute Configuration
Configuration
M1366 (R)-(ー)-2-Methoxy-2-(1-naphthyl)propionic Acid 1a 100mg 20,900 円 M1367 (S)-(+)-2-Methoxy-2-(1-naphthyl)propionic Acid 1b 100mg 20,900 円 光学活性化合物を誘導体化した後に,NMR スペクトルを測定して絶対配置を決定する 方法は,X 線結晶構造解析法,CD スペクトル法に比べて簡便な方法として利用されてい ます。本品 1(MαNP)は,光学活性アルコールの絶対配置を決定するための誘導体化試薬 です。これまでにも,MTPA Methoxy-2-trifluoromethyl-2-phenylacetic Acid),MNA(2-Methoxy-2-(2-naphthyl)acetic Acid),MPA (2-Methoxy-2-phenylacetic Acid) 等が,誘導体化 試薬として用いられていますが,ナフタレン環を有する 1 は,高い異方性効果と,キラル 炭素上に活性水素を持たないので誘導体化に際し,ラセミ化を起こさないという利点があ ります。例えば,(+)-2-Butanol の絶対配置は次のようにして決定することができます。 1)(+)-2-Butanol と 1a, 1b をそれぞれ反 応させてジアステレオマーを合成 2)得られた2つのジアステレオマーの 1H-NMRを測定 3)アルコール部分の化学シフトから ∆δ (=δR–δS)を求める(右表) 不斉中心に対してエステル部,プロトン の位置を図のように決め,∆δの値がプラ スになる基を右側に描くと,絶対配置を 決定することができます。従って,(+)-2-Butanolの絶対配置は右のようになり, S体と決定することができます。 また,(±)-2-Butanol と1aあるいは1bの一方から得られる2つのジアステレオマーは, シリカゲルHPLCで分離されるので光学活性アルコール等の光学純度を測定することもで きます。 文 献 1)東京化成工業(株), 特願 2000-115896.
2) N. Harada, M. Watanabe, S. Kuwahara, A. Sugio, Y. Kasai, A. Ichikawa,
Tetrahedron: Asymmetry, 11, 1249 (2000). + DCC, DMAP / CH2Cl2 OCH3 COOH CH3 OH OCH3 COO CH3 r.t., 24 h 1a C1 C2 C3 C4
NMR chemical shift data δR ∆δ
0.87 1.11 4.85 4.82 1.39 1.21 1.44 1.24 0.73 0.27 -0.24 +0.03 +0.18 +0.20 +0.46 C1 C2 C3 C3 C4 δS OMαNP H
LC-MS 用イオンペアー試薬 / Ion-Pair Reagents for LC-MS
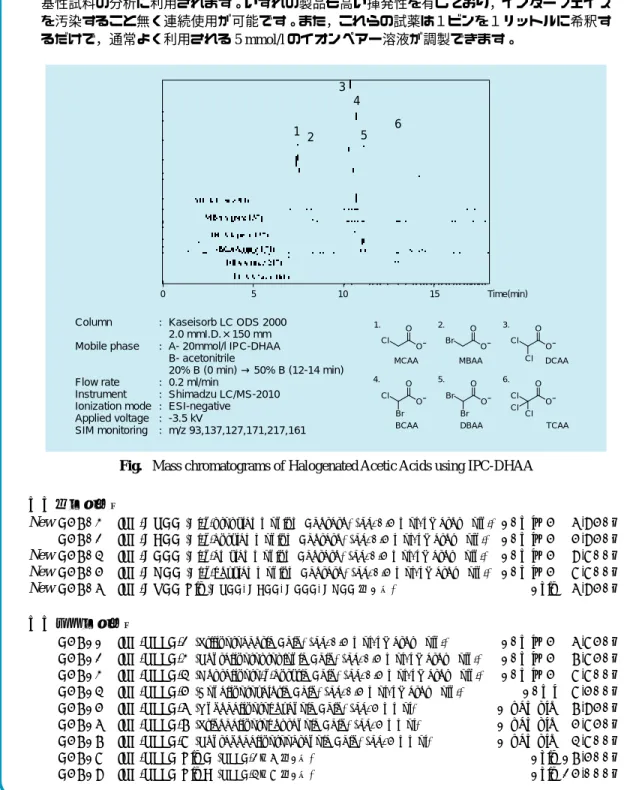
近年,HPLC の検出器として MS を組み込んだ LC-MS が盛んに利用され,そして分析対象物 質も増加しつつあります。今回紹介するジアルキルアンモニウムアセタート,ペルフルオロア ルカンカルボン酸は LC-MS 用イオンペアー試薬として開発された化合物で,それぞれ酸性,塩 基性試料の分析に利用されます。いずれの製品も高い揮発性を有しており,インターフェイス を汚染すること無く連続使用が可能です。また,これらの試薬は1ビンを1リットルに希釈す るだけで,通常よく利用される 5 mmol/l のイオンペアー溶液が調製できます。Fig. Mass chromatograms of Halogenated Acetic Acids using IPC-DHAA
酸性物質用
New A5703 IPC-DPAA (Di-n-propylammonium Acetate) (ca. 0.5 mol/l Water Soln.) 10ml×5 6,950円 A5702 IPC-DBAA (Di-n-butylammonium Acetate) (ca. 0.5 mol/l Water Soln.) 10ml×5 5,950円
New A5704 IPC-DAAA (Di-n-amylammonium Acetate) (ca. 0.5 mol/l Water Soln.) 10ml×5 9,800円
New A5705 IPC-DHAA (Di-n-hexylammonium Acetate) (ca. 0.5 mol/l Water Soln.) 10ml×5 8,800円
New A5706 IPC-DRAA Kit (DPAA, DBAA, DAAA, DHAA 各 1 本) 1kit 6,950円
塩基性物質用
A5711 IPC-PFFA-2 (Trifluoroacetic Acid) (ca. 0.5 mol/l Water Soln.) 10ml×5 6,850円 A5712 IPC-PFFA-3 (Pentafluoropropionic Acid) (ca. 0.5 mol/l Water Soln.) 10ml×5 7,850円 A5713 IPC-PFFA-4 (Heptafluoro-n-butyric Acid) (ca. 0.5 mol/l Water Soln.) 10ml×5 8,800円 A5714 IPC-PFFA-5 (Nonafluorovaleric Acid) (ca. 0.5 mol/l Water Soln.) 10ml 8,500円 A5715 IPC-PFFA-6 (Undecafluorohexanoic Acid) (ca. 5 mmol) 1 sample 7,950円 A5716 IPC-PFFA-7 (Tridecafluoroheptanoic Acid) (ca. 5 mmol) 1 sample 5,850円 A5717 IPC-PFFA-8 (Pentadecafluorooctanoic Acid) (ca. 5 mmol) 1 sample 4,800円
A5718 IPC-PFFA Kit A (PFFA-2∼6 各1本) 1kit 17,500円
A5719 IPC-PFFA Kit B (PFFA-4∼8 各1本) 1kit 25,000円
Column : Kaseisorb LC ODS 2000
2.0 mmI.D.×150 mm
Mobile phase : A- 20mmol/l IPC-DHAA
B- acetonitrile
20% B (0 min) → 50% B (12-14 min)
Flow rate : 0.2 ml/min
Instrument : Shimadzu LC/MS-2010
Ionization mode : ESI-negative Applied voltage : -3.5 kV SIM monitoring : m/z 93,137,127,171,217,161 Cl O O Br O O Cl O O Cl Cl O O Br Br O O Br Cl O O Cl Cl 1. 2. 3. 4. 5. 6.
MCAA MBAA DCAA
TCAA DBAA BCAA 1 2 3 4 5 6 0 5 10 15 Time(min)
ご照会は 学術部 TEL 03-5640-8857 FAX 03-5640-8868 E-mail [email protected] 弊社製品取扱店 または, 東京化成販売(株) TEL 03-3241-0573 FAX 03-3246-2094 大阪営業所 TEL 06-6228-1155 FAX 06-6228-1158 筑波サービスセンター TEL 0298-57-7825 FAX 0298-57-7845 ご注文は